
The European Molecular Biology Laboratory (EMBL) is an intergovernmental organization dedicated to molecular biology research and is supported by 28 member states, one prospect state, and one associate member state.
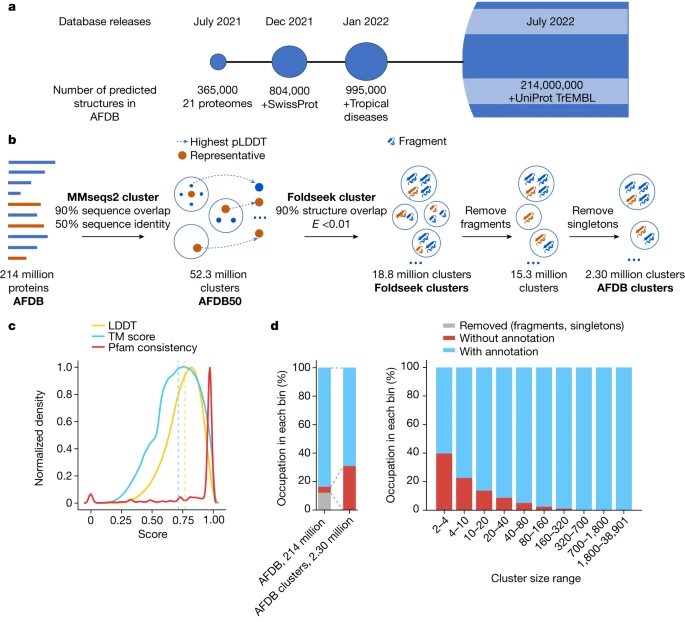
Using the AlphaFold database and a new algorithm called Foldseek Cluster, researchers have analysed over 200 million predicted protein structures, identifying unique evolutionary patterns.
The study uncovers new insights into the evolution of human immunity proteins by revealing structural similarities between human and bacterial proteins.
As the AlphaFold database continues to expand, algorithms such as Foldseek Cluster emerge as critical tools for navigating and interpreting the wealth of information made available by AI predictions.
By developing an efficient way to compare all predicted protein structures in the AlphaFold database, researchers have revealed similarities between proteins across different species. This work aids our understanding of protein evolution and has uncovered new insights into the origin of human immunity proteins.
The research was conducted by EMBL’s European Bioinformatics Institute (EMBL-EBI), the Institute of Molecular Systems Biology ETH Zurich, and the School of Biological Sciences Seoul National University.
The AlphaFold database is a transformative resource in the field of protein research, serving as a comprehensive repository of AI-predicted 3D structures for all known proteins. The database fills a critical gap in understanding protein function and evolution by offering high-quality structural predictions. Although AI predictions are not a substitute for experimentally determined structures, they do provide invaluable insights for the scientific community.
For this study, published in the journal Nature, the researchers developed a new algorithm known as Foldseek Cluster that can be used to analyze large sets of protein structures all at once. Foldseek Cluster was applied to the 200 million predicted protein structures in the AlphaFold database, identifying over 2 million unique structural clusters – groups of protein structures that are similar to each other in their three-dimensional shapes. One third of these clusters lack any previous annotations, meaning they had not before been described or categorized.
Structure-based clustering of the AFDB
The AFDB covers over 214 million predicted protein structures and has grown in several stages . The initial release focused on 20 key model organisms, while subsequent updates provided predictions for the Swiss-Prot dataset of the Universal Protein Resource18 (UniProt) and proteomes relevant to global health, taken from priority lists compiled by the World Health Organisation.
The current update covers most of the TrEMBL dataset of UniProt. The AFDB parses and archives these data and makes them accessible through bulk download options, programmatic access end points and interactive web pages. The programmatic access, in particular, facilitated the integration of AlphaFold models into other biological data repositories, such as Protein Data Bank Europe (PDBe)19, UniProt18, Pfam20, InterPro21 and Ensembl22.
Bridging the gap in protein science
Proteins are critical to processes that take place in the cell. Understanding protein structure is pivotal for studying their function and evolution. Despite significant advancements in sequence-based predictions of protein structures, computational limitations have made it difficult to study these structures at scale. Foldseek Cluster now enables structural comparisons and clustering at an unprecedented scale, reducing the time for such tasks by several orders of magnitude.
“We’ve entered a new era in structural biology where computational methods unlock unprecedented access to explore the protein universe,” said Martin Steinegger, Assistant Professor at the School of Biological Sciences Seoul National University. “We estimated that clustering all structures with established methods would have taken a decade when compared to the five days it took using our new method, Foldseek Cluster. Our algorithm can sift through millions of predicted protein structures in the AlphaFold database and cluster them based on their 3D shapes. This acceleration in computational power doesn’t just make things faster; it makes things possible.”
Protein evolution and immunity
The study also delves into the evolutionary implications of these clusters. While most clusters are ancient in origin, around 4% appear to be species-specific. This offers new insights into evolutionary phenomena such as de novo gene birth – when new genes arise from non-coding regions of the genome. The work also illustrates several examples of evolutionary relationships that could enrich our understanding of protein function across different species, including their role in human immunity.
“This work isn’t just about making comparisons more efficiently, it’s about gaining new insights into the evolutionary history of proteins,” said Pedro Beltrao, Associate Professor at the Institute of Molecular Systems Biology, ETH Zurich. “One of the most interesting findings from this study is our detection of structural similarities between human immune system proteins and those found in bacteria. This suggests that proteins involved in the immune system may have ancient evolutionary origins that we share with bacterial species. If true, this could reshape our understanding of immunity. Our research not only advances current knowledge but also lays out a roadmap for future investigations into the mysteries of protein function and evolution.”
Improving the AlphaFold database functionality
As the AlphaFold database and other life science databases continue to grow there is a significant need to help users sift through the vast amount of data while reducing the computational costs of analysing and managing these data. Approaches such as the Foldseek Cluster algorithm, that is scalable to billions of structures, will be invaluable in helping researchers navigate this wealth of information.
“Foldseek Cluster is more than just a technological advancement; it’s an enhancement that elevates the entire AlphaFold database experience for researchers worldwide,” said Sameer Velankar, Team Leader at EMBL-EBI. “With the explosion of predicted protein structures we have in AFDB, managing and navigating these data efficiently has been a significant challenge,” he continued. “Foldseek Cluster has revolutionized this process. We are working on integrating FoldSeek clusters into AFDB to streamline the analysis of large sets of protein structures and make it easier for our user community to find exactly what they’re looking for.”
AlphaFold and ESM (Evolutionary Scale Modeling) are both cutting-edge computational techniques used in the field of structural biology and protein folding. They have made significant advancements in our understanding of protein structures and functions.
AlphaFold:
AlphaFold is a deep learning-based method developed by DeepMind, a subsidiary of Alphabet Inc. It was first introduced in 2020 and gained widespread attention for its remarkable ability to predict protein structures with high accuracy.
The primary goal of AlphaFold is to predict the 3D atomic structures of proteins from their amino acid sequences. This is a critical task because the 3D structure of a protein is closely linked to its function, and understanding these structures can have profound implications for drug discovery and disease understanding.
AlphaFold uses deep neural networks and deep learning techniques, trained on a vast dataset of protein structures and sequences, to predict the 3D structure of a protein. It combines multiple sources of information, including sequence data and evolutionary information, to make highly accurate structure predictions.
AlphaFold’s breakthroughs have the potential to revolutionize the field of structural biology, as it can significantly accelerate the process of determining protein structures, which previously relied heavily on experimental techniques like X-ray crystallography and cryo-electron microscopy.
ESM (Evolutionary Scale Modeling):
ESM is another computational approach used for protein structure prediction, particularly focused on leveraging evolutionary information.
This technique is based on the idea that the evolutionary history of a protein, as reflected in its amino acid sequence and its homologous sequences found in different species, contains valuable information about its structure and function.
ESM models use deep learning architectures, similar to AlphaFold, but they emphasize the incorporation of evolutionary data, such as multiple sequence alignments, co-evolutionary patterns, and phylogenetic information.
By integrating this evolutionary information, ESM models can make accurate predictions about the 3D structure of a protein, often complementing the predictions made by other methods like AlphaFold.
Both AlphaFold and ESM have made significant contributions to the field of structural biology, enabling researchers to predict protein structures more accurately and efficiently. They are part of a broader effort to bridge the gap between genomics and functional biology, as understanding protein structures is crucial for deciphering their roles in various biological processes and for advancing fields like drug discovery and biotechnology.