
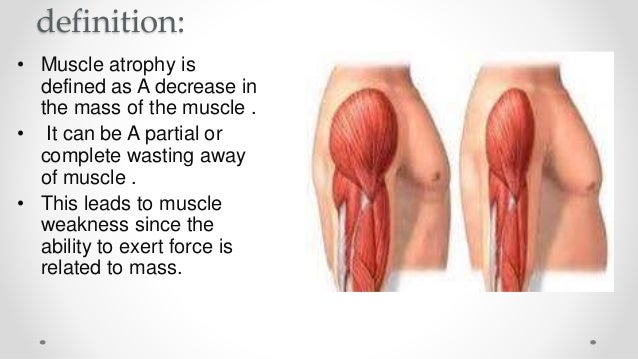
Muscle atrophy is defined as a decrease in the mass of the muscle; it can be a partial or complete wasting away of muscle, and is most commonly experienced when persons suffer temporary disabling circumstances such as being restricted in movement and/or confined to bed as when hospitalized. When a muscle atrophies, this leads to muscle weakness, since the ability to exert force is related to mass. Modern medicine’s understanding of the quick onset of muscle atrophy is a major factor behind the practice of getting hospitalized patients out of bed and moving about as active as possible as soon as is feasible, despite sutures, wounds, broken bones and pain.
Muscle atrophy results from a co-morbidity of several common diseases, including cancer, AIDS, congestive heart failure, COPD (chronic obstructive pulmonary disease), renal failure, and severe burns; patients who have “cachexia” in these disease settings have a poor prognosis. Moreover, starvation eventually leads to muscle atrophy.
Disuse of the muscles, such as when muscle tissue is immobilized for even a few days of unuse – when the patient has a primary injury such as an immobilized broken bone (set in a cast or immobilized in traction), for example – will also lead rapidly to disuse atrophy. Minimizing such occurrences as soon as possible is a primary mission of occupational and physical therapists employed within hospitals working in co-ordination with orthopedic surgeons.
The research picture has brightened considerably in the last decade for people with chromosome 5-related spinal muscular atrophy types 1 through 4, thanks to special genetic circumstances that provide researchers with unique opportunities for intervention.
Since 1995, scientists have known that a deficiency of functional SMN protein is the underlying cause of chromosome 5 SMA. (SMN stands for survival of motor neuron.)
Two nearly identical genes carry the genetic instructions for the SMN protein: SMN1 and SMN2. Proteins made from the SMN1 gene are full-length and functional, and appear to be necessary for the survival and proper function of the motor neurons. By contrast, proteins made using instructions from the SMN2 gene are shorter and tend to be less stable.
In SMA types 1 through 4, flaws (mutations) in each of the two copies of the SMN1 genes result in insufficient production of full-length, functional SMN protein.
Fortunately, a certain amount of full-length SMN protein can be made from the SMN2 gene. Many people have multiple copies of the SMN2 gene. These extra SMN2 genes can lessen the impact of a flaw in both SMN1 genes. In chromosome 5-related SMA, the more SMN2 genes a person has, the milder the course of SMA is likely to be.
Researchers are seeking to exploit this unique redundancy through development of various strategies that restore sufficient levels of the needed full-length SMN protein.
Since inception, MDA has contributed over $44 million dollars to SMA research.
Raising SMN levels through gene therapy
One research strategy to treat chromosome 5-related SMA types 1 through 4 is based on transferring SMN1 genes into the body to raise the level of full-length SMN protein. In a 2010 U.S. experiment, very young mice with an SMA-like disease received intravenous injections of genes containing instructions for the SMN protein, packaged inside modified type 9 adeno-associated viruses (AAV9 vehicles). The AAV9 vehicle reached its target — motor neurons in the spinal cord — and increased levels of SMN protein were subsequently found in the animals’ brains, spinal cords and muscles. The mice showed dramatic improvement of motor function and brain-to-muscle signaling, and a significant increase in survival. (This same gene delivery method later was successfully used in a monkey, although the monkey didn’t have an SMA-like disease. A similar method was used in a pig model of SMA as well, with promising results.)
Also in 2010, a British research group transferred SMN genes inside AAV9 delivery vehicles intravenously into mice with an SMA-like disease, improving the life span in these mice.
Brian Kaspar at Nationwide Children’s Hospital in Columbus, Ohio, discusses gene transfer in SMA in a May 2010 podcast. Dr. Kaspar, together with the biotechnology company AveXis, is developing this gene therapy approach for use in SMA patients. A phase 1 clinical trial testing whether intravenous delivery of AAV9-SMN could help type 1 SMA patients is ongoing.
Raising SMN levels using antisense oligonucleotides
Another strategy in development for SMA is using antisense oligonucleotides to cause more full-length SMN production from SMN2 genes. The SMN2 gene is similar in structure to the SMN1 gene. However, most of the protein made from the SMN2 gene is short and not functional. The antisense oligonucleotides alter how the SMN2 RNA is put together to increase the amount of full-length, functional SMN. In 2010, MDA-supported scientists announced that mice with an SMA-like condition showed a “robust and long-lasting increase” in full-length SMN protein in their spinal cords and in the motor neurons themselves after experimental antisense treatment. A February 2012 podcast features researcher Arthur Burghes discussing the antisense strategy.
Based off of laboratory research findings, ISIS Pharmaceuticals is developing an antisense oligonucleotide therapy called ISIS-SMNRx, which is currently in clinical trials. The antisense oligonucleotide has been administered in several trials to SMA patients including people with types 1, 2, and 3. Thus far, ISIS-SMNRx appears to be safe and well-tolerated and has shown encouraging results in some SMA patients in phase 2 trials. ISIS Pharmaceuticals is currently testing this drug in phase 3 clinical trials.

Genetic information moves from its storage form as DNA to a set of instructions known as RNA, from which protein molecules are made. Most of the RNA instructions from the SMN1 gene tell the cell to make full-length SMN protein. Most of the instructions from the SMN2 gene tell the cell to make short SMN protein.
Raising SMN levels using drugs
Several research strategies involve manipulating the genetic instructions provided by the SMN2 gene so that more full-length SMN protein can be made. The SMN2 gene is similar in structure to the SMN1 gene. However, most of the protein made from the SMN2 gene is short and not functional. This research approach uses small molecule drugs that target the SMN2 gene to change how the SMN2 RNA is put together, with the goal of increasing production of full-length, functional SMN or to increase the overall level of SMN through other means.
Groups of pharmaceutical companies are testing different drugs that act through this approach in SMA patients. Novartis is beginning a phase 2 trial in type 1 SMA patients to test their drug called LMI070. Additionally, Roche, together with PTC Therapeutics, initiated a phase 2 clinical trial to test their drug RG7800 in SMA patients. With MDA support, a company called Repligen has developed an experimental compound called RG3039 (also called quinazoline), which is designed to interfere with an enzyme and thereby increase production of full-length SMN protein from the SMN2 gene. This drug is currently being tested in a phase 1 clinical trial in healthy volunteers.
Other companies developing drugs to increase levels of full-length SMN currently are in the preclinical development stage. Paratek Pharmaceuticals is working on a small molecule compound that is similar to an antibiotic called tetracycline. Additionally, the California Institute for Biomedical Research (CALIBR) is in the process of optimizing drugs that upregulate SMN levels through a different mechanism than RNA splicing.
X-linked SMA research
An X chromosome gene has been identified that, when mutated, causes X-linked SMA. The gene codes for theUBE1 protein, which is part of a cellular waste disposal system. Without functional UBE1 protein, this important waste disposal system probably malfunctions.
Scientists are now studying the UBE1 gene and protein with an eye to identifying therapeutic targets in X-linked SMA.
Targeting muscle
Motor neurons are a specialized type of nerve cell that dies in people with SMA. These motor neurons are the wires that connect the brain and spinal cord to the muscles, and their death leads to muscle weakness and paralysis in SMA. One approach researchers are pursuing for SMA focuses on protecting muscles from paralysis and increasing their strength. Although this approach does not fix the underlying genetic problem in SMA, drugs that enhance muscle function could likely be used in combination with other therapies that act on the SMN genes.
Cytokinetics is developing drugs that increase the ability of the muscle to contract. These drugs have shown early promise in patients with a similar motor neuron disease called amyotrophic lateral sclerosis (ALS). Together with Astellas, Cytokinetics is developing a similar drug called CK-2127107 for SMA. This drug has been tested in a phase 1 clinical trial of healthy volunteers, where it proved to be safe and able to increase muscle force. Cytokinetics is planning to test CK-2127107 in a phase 2 clinical trial in SMA patients.
Newborn screening
Most evidence suggests that treatment of babies with type 1 SMA should be done as early in life as possible, and that it may be necessary to start screening for the disease in newborns to identify them early. Newborn screening is performed for many diseases and could be one means to identify and treat infants with SMA before they lose motor function. There are efforts underway to conduct pilot studies in certain areas to assess the feasibility of newborn screening for SMA. If a robust treatment for SMA becomes approved in the US, newborn screening for the disease may be pursued for all babies.
Chromosome 14-related (DYNC1H1) research
Flaws in the cytoplasmic dynein 1 heavy chain 1 (DYNC1H1) gene on chromosome 14 can lead to a rare form of SMA called SMA-LED, which predominantly affects muscles in the legs. The DYNC1H1 gene works as a “motor” to transport cellular components. Mutations in the DYNC1H1 gene result in disruptions to the motor’s function. Much work is yet to be done on this newly discovered cause of SMA.
Neuroprotection
Motor neurons are the nerve cell type that degenerates in SMA, leading to muscle weakness and paralysis. While some research is focused on strategies to increase SMN levels to help motor neurons, other scientists are focusing on broad neuroprotection. This research aims to prevent motor neurons from becoming dysfunctional and dying rather than altering the genetics of the SMN genes. Neuroprotective strategies could likely be used in combination with other drugs that address the underlying genetic problem in SMA.
In 2007, the French company Trophos identified a novel compound called olesoxime (TRO19622) that was able to protect motor neurons from death in a dish. Since then, olesoxime has been tested in SMA patients in clinical trials with encouraging results. A phase 2 trial conducted on types 2 and 3 SMA patients in Europe showed that olesoxime preserved motor function and seemed safe and well-tolerated. Trophos, which was acquired by Roche in 2015, has planned to file a New Drug Application (NDA) with the FDA in the US in order to move olesoxime forward.
Biomarkers for SMA
For many diseases, there are indicators in the body that change when a person has a disease or when the disease gets worse or better. These indicators are called “biomarkers” and can be found by testing the blood or urine, or by using other more sophisticated types of testing, such as nerve conduction tests commonly used to diagnose motor neuron disorders. One area of active research within the SMA field is to identify biomarkers which could be used to tell how SMA patients are progressing and whether they are responding to a potential treatment. Since motor function tests can be variable and difficult to conduct, especially in young infants, identification of biomarkers would be useful in the efforts to find treatments for SMA.
SMA biomarkers are currently being evaluated in the NIH NeuroNEXT clinical trial, titled “SMA Biomarkers in the Immediate Postnatal Period of Development.” In this trial, infants with SMA and healthy controls are being followed over time, and a variety of testing is being conducted to determine whether any reliable biomarkers can be identified for SMA.
Credit: Muscular Dystrophy Association